Icterícia: Um alerta amarelo!
")
- Introdução

Como a icterícia é comumente observada nos recém-nascidos (60% a 80% dos RN apresentam icterícia na primeira semana de vida), não é raro que este seja um sinal pouco valorizado nesta faixa etária, o que contribui para o encaminhamento tardio do paciente colestático. Assim, é de extrema importância que o Pediatra fique atento à duração da icterícia nos primeiros dias de vida e à coloração das fezes e da urina. Este procedimento simples pode fazer a diferença entre a sobrevida ou não do paciente. Se as fezes estiverem claras, a urina escura e/ou a icterícia ultrapassar 14 dias de vida, a criança deve ser avaliada do ponto de vista clínico e laboratorial, como citado em trabalho de revisão por De Bruyne et al, 2011.
No Brasil, ainda hoje, os pacientes são, em sua maioria, encaminhados tardiamente (Carvalho E et al, 2010). A campanha do Alerta Amarelo busca, com a divulgação destes conhecimentos, uma nova era no manejo da colestase do neonato e do lactente no Brasil!
- Icterícia: diagnóstico diferencial
A icterícia pode ocorrer por aumento da produção da bilirrubina, diminuição de sua captação, redução da conjugação pelos hepatócitos e/ou diminuição da sua secreção. Os três primeiros processos cursam com elevação da bilirrubina não conjugada (bilirrubina de reação indireta – BI), enquanto a diminuição da secreção biliar apresenta-se com aumento da bilirrubina conjugada (bilirrubina de reação direta – BD). Assim, definir se a condição é decorrente do aumento da BI ou da BD é o primeiro passo para o diagnóstico diferencial do paciente ictérico. A icterícia com predomínio da BI pode ser decorrente do aumento de sua produção, diminuição da sua captação e/ou conjugação pelo hepatócito. A distinção entre estes processos pode ser realizada pelo nível dos reticulócitos e representa a segunda etapa do diagnóstico diferencial do paciente ictérico. A Figura 1 ilustra o algoritmo do diagnóstico diferencial do neonato e do lactente ictérico e demonstra que, com dois exames de baixo custo e fácil disponibilidade (dosagem sérica de bilirrubinas e reticulócitos), é possível definir o processo responsável pela icterícia e elaborar o diagnóstico diferencial do caso em questão, estabelecendo se é colestático ou não.
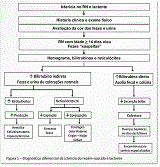
- Colestase neonatal
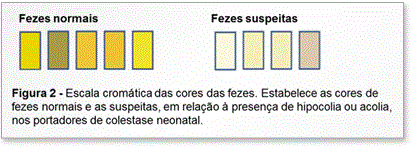
Do ponto de vista clínico, a colestase se manifesta por icterícia, hipocolia ou acolia, colúria, prurido e xantomas. Destas manifestações, a icterícia é o sinal que mais chama a atenção do clínico, mas enfatizamos que analisar a coloração das fezes e da urina faz parte da avaliação do paciente ictérico, pois auxilia no diagnóstico diferencial entre os casos colestáticos e não colestáticos. Em relação à cor das fezes, a detecção da hipocolia ou acolia pode contribuir para o diagnóstico precoce dos pacientes colestáticos e, por este motivo, no Brasil foi incluída na Caderneta de Saúde da Criança, distribuída pelo Ministério da Saúde, a escala colorimétrica das fezes. Esta escala define as fezes normais e as suspeitas por um sistema de graduação de cores, como ilustrado na Figura 2.
Do ponto de vista laboratorial, o diagnóstico de colestase é sugerido pela presença de BD >1 mg/dL se o nível de BT for <5 mg/dL e de BD >2 mg/dL ou maior que 20% do total se BT >5 mg/dL. Além da elevação da BD, observa-se aumento das enzimas canaliculares, da gamaglutamiltransferase (GGT) e da fosfatase alcalina (FA), das concentrações séricas dos ácidos biliares e do colesterol. Na avaliação histopatológica, observa-se acúmulo de pigmento bilirrubínico nos hepatócitos e canalículos biliares, além de outros achados relacionados à etiologia do caso.
Após a identificação da icterícia colestática, procede-se à investigação do diagnóstico etiológico. O diagnóstico diferencial é cada vez mais amplo e heterogêneo, pois várias doenças vêm sendo descobertas nas últimas décadas. Nesta etapa, devem ser identificadas, especialmente, as situações que ameaçam a vida e que têm possibilidade de tratamento, como a atresia biliar, as doenças infecciosas, as genéticas e metabólicas (galactosemia, tirosinemia tipo I, o erro inato do metabolismo dos sais biliares e a fibrose cística) e as endocrinopatias (hipopituitarismo). Atualmente, a atresia biliar continua sendo responsável por 25% dos casos de colestase neonatal, sendo o primeiro passo definir se o tratamento será clínico ou cirúrgico.
Na exploração diagnóstica, apesar dos avanços dos métodos complementares diagnósticos, a história
fundamentais. Os achados variam de apenas icterícia leve até os sinais de insuficiência hepática grave. Na atresia biliar (forma perinatal) a criança, em geral, teve bom peso de nascimento, apresenta-se com icterícia, acolia e colúria, sem outras alterações do exame físico. Já na forma embrionária da atresia biliar (20 a 30% do total), os lactentes podem apresentar poliesplenia, cardiopatia congênita, má rotação intestinal, situs inversus ou outras malformações congênitas. Os RN com baixo peso ao nascimento, microcefalia, púrpura e coriorretinite devem ser submetidos à pesquisa de infecções congênitas. Alterações dismórficas são observadas nas cromossomopatias. A irritabilidade, os vômitos, a letargia, os sinais de hipoglicemia e a acidose metabólica chamam a atenção para os erros inatos do metabolismo. As anormalidades neurológicas são observadas na síndrome de Zellweger, nas mitocondriopatias e como consequência de complicações como os episódios de hipoglicemia, hiperamonemia e hemorragia intracraniana.
Embora os transtornos que levam à insuficiência hepática fulminante sejam incomuns, um alto grau de suspeição deve ser mantido, já que o diagnóstico precoce e o início imediato do tratamento são a única esperança de sobrevivência, como ocorre nos casos de galactosemia, tirosinemia tipo I e hemocromatose neonatal.
Quanto aos exames complementares, como o diagnóstico diferencial é amplo, devem ser solicitados conforme a principal suspeita, direcionada pelos achados clínicos. De modo geral, os exames têm dois objetivos diferentes. O primeiro é avaliar a presença de colestase e definir a gravidade da doença e do acometimento hepático. O segundo consiste em definir o diagnóstico etiológico. A Figura 3 ilustra o raciocínio diagnóstico frente ao lactente colestático.
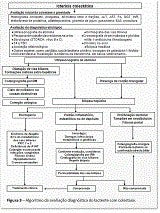
- Experiência brasileira
Uma pesquisa que avaliou uma série grande de crianças com atresia biliar (Carvalho et al, 2010) de todas as regiões brasileiras e avaliou os dados relativos à portoenterostomia, ao transplante hepático e à sobrevida constatou que atualmente, no Brasil, os pacientes, em sua maioria, não são operados ou são submetidos à portoenterostomia tardiamente, após 60 dias de vida, situação que influencia negativamente sua sobrevida com fígado nativo (sem transplante hepático). A idade observada, no momento da portoenterostomia, foi superior à desejável, revelando o encaminhamento tardio, um problema a ser resolvido em nível nacional, independentemente da região ou da categoria da cidade, seja capital ou cidade do interior.
Experiências de outros países têm demonstrado que o encaminhamento destes pacientes pode ser melhorado por meio do aprimoramento nas práticas médicas e pela adoção de políticas de saúde. A exemplo destes países, no Brasil algumas medidas estão sendo adotadas para aumentar a conscientização da sua população para a importância do diagnóstico precoce da colestase neonatal, utilizando sinais de alerta e sistemas de triagem, como a campanha do Alerta Amarelo e o uso da escala colorimétrica das fezes para identificar a acolia (Figura 2).
- Tratamento
A abordagem terapêutica da colestase neonatal tem como objetivos evitar as consequências da colestase independentemente da etiologia e instituir o tratamento de doenças específicas, se disponível.
Quanto às consequências da colestase crônica, elas são graves e independem da etiologia. A retenção dos constituintes da bile e a redução da concentração intraluminar dos sais biliares desencadeiam uma série de eventos, como a icterícia, o prurido, a deficiência de vitaminas, a desnutrição, além de contribuir para a progressão da doença hepática. Assim, quanto ao tratamento inespecífico, a nutrição adequada, a suplementação com vitaminas lipossolúveis e o uso de agentes coleréticos podem minimizar os efeitos adversos da colestase crônica. Lembramos que em alguns lactentes colestáticos as consequências da deficiência das vitaminas lipossolúveis podem ser os únicos sinais/sintomas da presença de colestase. Nestas crianças a hiperbilirrubinemia pode não ser clinicamente evidente e o atraso no diagnóstico pode causar graves problemas, como a hemorragia intracraniana decorrente da deficiência de vitamina K, como demonstrado por Ho et al (2012). Assim, as coagulopatias devem ser investigadas nos lactentes com hiperbilirrubinemia de reação direta, e a administração imediata de vitamina K deve ser instituída.
O tratamento específico das doenças deve levar em consideração que algumas opções terapêuticas são tempo-dependentes e que determinados fármacos podem ser eficazes ou adequados apenas num estágio da doença, como no uso do NTBC [2-(2-nitro-4-fluoromethylbenzoyl)- 1,3-cyclohexanedione] para a tirosinemia tipo I. Outros exemplos constituem a dietoterapia, para a galactosemia, e a portoenterostomia, para a atresia biliar.
- Considerações finais
A campanha do Alerta Amarelo orienta que todo recém-nascido que persistir com icterícia com idade igual ou maior que 14 dias deve ser avaliado do ponto de vista clínico (global e coloração de fezes e urina) e laboratorial (bilirrubinas). Se as fezes forem “suspeitas” e/ou o aumento de bilirrubina for da reação direta, a criança deve ser encaminhada para serviços especializados. Este procedimento simples pode fazer a diferença entre a sobrevida ou não de algumas crianças!
Veja mais detalhes nos links de acesso: http://www.ncbi.nlm.nih.gov/pubmed/21249394 http://www.ncbi.nlm.nih.gov/pubmed/21140036 http://www.ncbi.nlm.nih.gov/pubmed/23039903
Artigos de referência:
- De Bruyne R, Van Biervliet S, Vande Velde S, Van Winckel M. Clinical practice: neonatal cholestasis. Eur J Pediatr. 2011 Mar;170(3):279-84. Epub 2011 Jan 20. Review. PubMed PMID: 21249394.
- Carvalho E, Santos JL, Silveira TR, Kieling CO, Silva LR, Porta G, Miura IK, De Tommaso AM, Brandão MÂ, Ferreira AR, Macêdo JR, Almeida Neto JT; Grupo de Estudos em Hepatologia Pediátrica do Brasil. Biliary atresia: the Brazilian experience. J Pediatr (Rio J). 2010 Nov-Dec;86(6):473-9. PubMed PMID: 21140036.
- Ho SS, Haller W, Catto-Smith AG. Yellow is pale: The complications and challenges of late diagnosis of extrahepatic biliary atresia. J Paediatr Child Health. 2012 Oct 8. doi: 10.1111/j.1440-1754.2012.02501.x. [Epub ahead of print.]
Se gostou deste post, também poderá gostar de